Encefalopatias
espongiformes en el ser humano
Dr. Marcial García Rojo
Servicio de Anatomía Patológica. Complejo Hospitalario de Ciudad Real
Resumen
Introducción
Qué son los priones
¿Cómo puede una proteína expresada normalmente
ser también un patógeno transmisible?
¿Cómo
se convierte PrPC en PrPSc?.
La
proteína prión en humanos.
Incidencia
Encefalopatía
Espongiforme Bovina.
Enfermedad
de Creutzfeldt Jacob clásica.
Nueva
variante de ECJ.
Características clínicas de enfermedades priónicas
en humanos
Enfermedad
de Creutzfeldt-Jakob esporádica (ECJ)
Forma
familiar de ECJ.
Forma
Iatrogénica de ECJ.
Enfermedad
de Gerstmann-Straussler-Scheinker (GSS)
Insomnio
fatal (IF)
Variante
de la ECJ.
Tratamiento de las enfermedades por priones
Genética de las encefalopatías espongiformes
transmisibles
Transmisión de EET en humanos
Transmisión
por derivados de la sangre.
Medidas a tomar
Perspectivas de futuro
Información en Internet
Bibliografía consultada
Resumen
Las Encefalopatías Espongiformes Transmisibles (EET) son enfermedades
raras, con periodos de incubación largos (meses a años), que afectan al
sistema nervioso central y progresan lentamente hasta la muerte del
enfermo. En los animales, la EET más conocida actualmente es la
encefalopatía espongiforme bovina (EEB) o "mal de las vacas
locas". En el ser humano de conocen este tipo de enfermedades desde
hace más de 80 años y dan lugar a cuadros clínicos conocidos como
enfermedad de Creutzfeldt-Jakob, kuru, insomnio fatal, etc. A pesar de su
rareza en el ser humano, las enfermedades por priones han adquirido una
importancia social elevada debido, por una parte, a la posible conexión
entre una nueva variante de enfermedad de Creutzfeldt-Jakob (vECJ)
que se describió en 1995 en humanos y la encefalopatía espongiforme
bovina. En las EET hay bastantes evidencias a favor de que el agente
causal sea una proteína resistente a proteasas (prión) y no un virus,
que sorprendentemente existe normalmente en el huésped, con una
estructura algo distinta ("prión sano"), aunque hay evidencias
que indican que otras proteínas (Dpl o doppel) también podría
participar en la patogenia de estas enfermedades.
Hasta el momento,
se han declarado 182.507 reses enfermas de EEB en todo el mundo, la mayoría
(179.441) en el Reino Unido, y se han confirmado 13 casos en España. Por
otra parte, se han observado 91 pacientes afectados de vECJ,
principalmente en el Reino Unido. La importancia de la aparición de la
vECJ es que el agente causal ha sido capaz de saltar la llamada
"barrera de las especies". Aunque experimentalmente se ha
transmitido en animales, no hay evidencias directas de transmisión de
vECJ a través de transfusión sanguínea o vacunas en humanos.
Desconocemos el número de casos de vECJ que aparecerán en los próximo años,
pero conviene destacar que la incidencia de nuevos casos en el Reino Unido
ha permanecido baja durante estos años.
Introducción
Las enfermedades por priones, también denominadas Encefalopatías
Espongiformes Transmisibles (EET), son una familia de enfermedades
neurodegenerativas raras, con periodos de incubación largos, lentamente
progresivas, y universalmente fatales, que afectan a personas y animales(1).
La encefalopatía
espongiforme bovina (EEB) o "mal de las vacas locas", de la que
se han detectado más de 180.000 reses enfermas desde su detección por
primera vez en Reino Unido en 1986(2),
pertenece a este grupo de enfermedades, junto con el "scrapie",
conocida desde hace unos 250 años. El scrapie sólo afecta a las ovejas y
cabras(3)
su nombre se debe a que la enfermedad se caracteriza por un intenso
prurito que obliga a rascarse (scraping en inglés) a las ovejas,
junto con ataxia(4).
La "enfermedad
de Creutzfeldt-Jakob" (ECJ), conocida desde 1920, que afecta al ser
humano, también pertenece a este grupo de enfermedades. En 1957 se
describió la afectación de seres humanos por una enfermedad similar,
llamada kuru, en caníbales de Papúa (Nueva Guinea)(5).
La gran
variabilidad de presentación clínica de estas enfermedades por priones
requieren otros criterios más objetivos, como los hallazgos anatomopatológicos,
para poder clasificar las diversas enfermedades. De esta forma, además de
la ECJ y kuru, otras formas descritas de enfermedades priónicas humanas
incluyen la enfermedad de Gerstmann-Straussler-Scheinker (GSS) el Insomnio
Fatal (IF). En los últimos años se han descrito una variedad de cuadros
clínicos de etiopatogenia priónica que han ampliado el espectro clínico
de estas enfermedades en humanos: demencia por prión sin patología
característica, demencia con paraparesia espástica, Encefalopatia
Espongiforme Familiar asociada a nueva mutación en el gen PrP, Gliosis
Subcortical Progresiva, o enfermedad mental sin signos neurológicos. Así
mismo, casos descritos como enfermedad de Alzheimer familiares han sido
reexplorados y catalogados de origen priónico(6).
A pesar de su
rareza en el ser humano, las enfermedades por priones han adquirido una
importancia social elevada debido, por una parte, a la posible conexión
entre una nueva variante de enfermedad de Creutzfeldt-Jakob que se
describió, en 1995, en humanos (que abreviaremos como vECJ) y la
encefalopatía espongiforme bovina, y por otra parte, por la posibilidad
de contaminación de la sangre humana y de productos derivados de la
sangre (como el plasma) por el agente de la vECJ(3).
Otras enfermedades priónicas descritas en animales incluyen la
encefalopatía transmisible del visón, la enfermedad devastadora crónica
del ciervo y el alce (chronic wasting disease ó CWD), y la
encefalopatía espongiforme felina, entre otras. La mayoría de estas
enfermedades, incluyendo la EEB, se cree que son debidas a la ingestión
de productos animales contaminados con scrapie de oveja (figura 1), aunque
la CWD parece ser una enfermedad de aparición natural en Norteamérica(4).
Todas estas
enfermedades tiene dos características comunes: su naturaleza
transmisible y sus características anatomopatológicas, incluyendo la vacuolización
marcada de la sustancia gris del cerebro, que origina un aspecto en
esponja cuando se observa en el microscopio óptico; por ello, estas
enfermedades tiene en nombre común de encefalopatías espongiformes
transmisibles (EET).
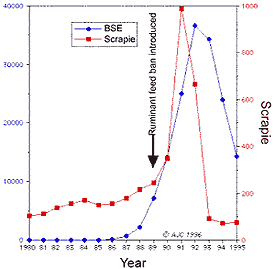 La EEB se caracteriza en los animales por nerviosismo, alta reactividad a
estímulos externos y dificultad de movimientos, sobre todo en las
extremidades posteriores. La edad media de presentación de los síntomas
es de 4 a 5 años(7).
La EEB se caracteriza en los animales por nerviosismo, alta reactividad a
estímulos externos y dificultad de movimientos, sobre todo en las
extremidades posteriores. La edad media de presentación de los síntomas
es de 4 a 5 años(7).
El problema que
plateamos en este escrito es si la enfermedad de las "vacas
locas" o encefalopatía espongiforme bovina puede ser transmitida al
ser humano y qué consecuencias tendría para el ser humano la infección
por los priones de la encefalopatía espongiforme bovina(3).
En octubre de 1995,
se produce un hecho crítico, cual es la publicación por Bateman y
Britton y cols, como cartas al director de la revista Lancet de los dos
primeros casos de lo que se denominará más tarde Enfermedad de
Creutzfeldt-Jakob nueva variante (vECJ)(8,9).
Los dos pacientes tenían en común algunas características clínicas que
les diferenciaba de la ECJ clásica, sobre todo la edad de presentación,
las manifestaciones clínicas (trastorno neuropsiquiátrico progresivo que
conduce a ataxia, demencia y mioclonias o corea), la duración y la
ausencia del EEG típico. Más tarde, en abril de 1996 Will y cols
publican ya una serie de 10 casos de esta vECJ y apuntan a la posibilidad
de una relación con la EEB como causa de los mismos(10).
La variante de la
enfermedad de Creutzfeldt-Jakob, descrita casi exclusivamente en el Reino
Unido, a diferencia de la descripción original de la enfermedad de
Creutzfeldt-Jakob clásica, se ha descrito también en adolescentes y
tiene unas características histopatológicas (marcado cambios
espongiformes y numerosas placas floridas por todo el cerebro) que la
distinguen de la enfermedad de Creutzfeldt-Jakob clásica (11,12).
Los estudios de
laboratorio, incluyendo los experimentos de transmisión en ratones,
proporcionan fuertes evidencias que apoyan la hipótesis que indica que la
vECJ está causada por la encefalopatía espongiforme bovina(12).
Qué
son los priones
Prusiner propuso que una fracción proteica era el origen de estas
enfermedades e introdujo el término prión para enfatizar su
naturaleza tanto proteica como infecciosa(13).
Aunque ni la teoría viral ni la teoría proteica han sido probadas, hay
bastantes evidencias a favor de que se trate sólo de una proteína y no
de un virus, por lo que Prusiner obtuvo el premio Nobel de Medicina en
1997.
Se sospechó la
naturaleza proteica de este agente infeccioso porque la exposición a
radiaciones ionizantes y ultravioleta degradan los ácidos nucleicos pero
no reducen la infectividad de las fracciones de scrapie experimentalmente,
mientras que los procedimientos que degradan las proteínas, como la
exposición prolongada a proteasas, se correlacionan con un menor poder
infeccioso(14).
En los cerebros de animales con encefalopatías espongiformes
transmisibles se aisló una proteína relativamente resistente a proteasas
(figura 2), lo sorprendente fue comprobar que esta proteína se codificaba
normalmente por genes de los cromosomas del huésped(15).
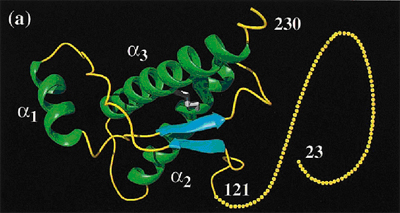 Fig. 2.
Estructura tridimensional de la proteína priónica bovina (7).
Fig. 2.
Estructura tridimensional de la proteína priónica bovina (7).
¿Cómo
puede una proteína expresada normalmente ser también un patógeno
transmisible?
Los priones son
proteínas complejas (cadenas de polipéptidos), cuya estructura se conoce
mejor cada día; la proteína del prión se conoce como PrP.
Existen dos isómeros
principales de la proteína PrP: la forma celular o no patógena,
denominada PrPC, y la forma patógena o forma inductora del
scrapie, denominada PrPSc. Tanto PrPC como PrPSc
tienen la misma secuencias de aminoácidos, pero difieren en sus
propiedades (tabla 1)(4).
Tabla
1. Propiedades de la Proteína de prión sano (PrPC) y
patológica (PrPSc)
|
|
|
Característica
|
PrPC
|
PrPSc
|
Bioquímicas
|
|
|
Solubilidad
en detergentes no desnaturalizantes
|
Soluble
|
Insoluble
|
Degradación
por proteasas
|
Total
ðÕ��p¡�� ’��Ð�
�P·��€'��À™��à¨��€���p��� ������� ���€���°����Q��n="left" width="129"> |
|
Forma
|
Predominantemente
helicoidal
|
Al
menos un 40% de su estructura es en hoja plegada
|
¿Cómo
se convierte PrPC en PrPSc?
Algunos autores
denominan prión sólo a la forma alterada de la proteína celular
funcional que ha perdido su función normal pero que ha adquirido la
capacidad de transformar la forma normal en patológica.
Los priones son
infecciosos a pesar de carecer de ácidos nucleicos con información genética.
La transmisión se cree que se realiza mediante la conversión de una
proteína normal en el huésped en una forma similar patológica(17).
Los mecanismos que
posiblemente inician la conversión de PrPC a PrPSc
incluyen a) mutaciones de línea germinal del gen de la proteína priónica
humana (PRNP), b) una mutación somática dentro de una neurona
determinada, y c) conversión espontánea del PrPC en una
conformación aberrante que no se pliega de forma apropiada en su
estructura nativa(4).
Independientemente
del mecanismo de inicio, una vez que se ha generado una "unidad
infecciosa", PrPSc parece actuar como una plantilla
conformacional mediante la cual PrPC se convierte en una nueva
molécula de PrPSc mediante interacción entre las proteínas
de PrPC y PrPSc (4).
De hecho, se ha observado que los ratones a los que se les ha eliminado el
gen normal PrP no desarrollan la enfermedad por priones tras la inoculación
con scrapie(18).
Además, los ratones transgénicos (Tg) que expresan un gen quimérico (de
dos especies distintas) hecho de segmentos humanos (Hu) y de ratón (M),
llamados Tg(MHu2M), desarrollan priones PrPSc quiméricos ratón-humanos
proteasa-resistentes en sus cerebros, cuando son inoculados con
extractos de cerebro de humanos con enfermedad por priones(19).
Por lo tanto, los
priones no se auto-replican sino que se convierten de PrPC no
patógenos a PrPSc patógenos.
La
proteína prión en humanos
En los humanos,
el gen de la proteína priónica humana, PRNP, se localiza en el cromosoma
20 y codifica una PrP de 253 aminoácidos(). Esta PrP es
glucosilada en dos lugares unidos a la asparagina y se une a la superficie
celular mediante un gancho glucosil-fosfatidil-inositol (GPI)(4).
La PrP es regulada
durante el desarrollo y constitucionalmente se expresa normalmente en el
adulto. El mayor nivel de expresión de PrP se identifica dentro de las
neuronas, pero también se identifican niveles más bajos en otros tejidos
periféricos como pulmón, corazón, riñón, páncreas, testículos,
leucocitos sanguíneos y plaquetas. Inmunohistoquímicamente se ha
detectado PrPC también en la unión neuromuscular. También se
ha descrito el transporte neuronal periférico y central anterógrado de
PrPC (4).
La función normal
del PrPC no se conoce, pero hay datos que sugieren que
participe en la función sináptica y en la unión al cobre.
Experimentalmente, cuando en los ratones en los que el gen PrP es
deleccionado, en algunos estudios no se observan anomalías de desarrollo
o del comportamiento, y otros muestran ligeras alteraciones del ritmo
circadiano o ataxia(4). La causa de esta ataxia podría ser la
sobreexpresión de un gen próximo al PRNP, denominado PRND, que produce
una proteína llamada Dpl ó doppel ("doble") que es homólogo
en un 25% con el extremo carboxilo del PrP de ratón().
Aunque los
principales cambios patológicos aparecen en el encéfalo, el agente
infeccioso pronto se acumula en el tejido linfoide. La aparición o no de
enfermedad clínicamente manifiesta depende de la presencia de un sistema
inmune intacto, incluyendo linfocitos B maduros(17).
Collinge y
colaboradores, en octubre de 1996, publicaron los primeros análisis
moleculares de la Proteina Priónica (PrP), en concreto los patrones de
glucosilación de la misma mediante Western blot. El Western blot de la
PrP de extractos de cerebro infectado por priones revela 3 bandas mayores,
que se corresponden a PrP que tienen una, dos o ninguna cadena de
polisacaridos unidos a la parte amino-terminal. En los cerebros con
priones, el tratamiento con esta proteasa, condiciona un incremento de la
movilidad de estos 3 tipos de moléculas debido a un aumento en las
roturas de las terminales amino. En la ECJ esporádica aparecen dos
patrones de movilidad, denominados tipo 1 (corresponde con los homocigotos
para Met) y 2 (homocigotos para Val), en la ECJ iatrógena por vía periférica
presenta un patrón nuevo tipo 3 y en la vECJ y la EEB presenta un patrón
también diferente, tipo 4. En animales infectados con material de EEB se
mantenía este patrón de glucosilación (tipo 4), que corresponde a una
alta proporción de PrP biglucosilada. La conclusión fue que se trataba
posiblemente de la misma cepa de prión().
No se conoce aún
el mecanismo por el que las células mueren al generarse los priones. El
simple acúmulo de proteína patógena parece no ser suficiente para
explicar la enfermedad, aunque debe ser un factor esencial en la disfunción
celular(4).
Algunos estudios
indican que el diagnóstico de vECJ podría confirmarse mediante biopsias
de amígdala palatina, pues hasta el momento sólo es posible realizarlo
en biopsias cerebrales o en el estudio autópsico().
La detección de la
PrP de la CJD es hoy el marcador diagnóstico más fiable de las
enfermedades por priones. Puede ser detectada mediante técnicas de
Western blot, histoblot, microscopía electrónica, pero tienen el
inconveniente de que requieren material fresco lo cual disminuye su uso
debido a la capacidad infectiva de la proteína priónica().
Otra posibilidad es
la detección mediante técnicas de inmunohistoquímica realizadas sobre
material fijado en formol e incluído en parafina, al que se ha realizado
un paso previo por ácido fórmico para anular la infectividad. PrPC
es una proteína frágil que puede ser preservada sólo con procedimientos
especiales de inclusión, mientras que la PrP de la CJD parece ser más
resistente a la fijación en formol e inclusión en parafina; y aunque los
mecanismos moleculares de los sistemas de recuperación antigénica no son
bien conocidos, parece ser que éstos pretratamientos, destruyen la PrPC
con lo que no interferiría en la detección de la PrP de la CJD. Aunque
recientemente se ha desarrollado un anticuerpo que sólo reconoce la
isoforma alterada(24).
El proceso biológico de
propagación de un prión se inicia con la interacción de la proteína
priónica anómala exógena (PrPSc o PrPCJD) con la
proteína priónica "sana" (PrPC) o con una forma
parcialmente desnaturalizada de ésta. Por otra parte, mutaciones
puntuales y variaciones en la longitud de la cadena polipeptídica de la
proteína priónica "sana" así como alteraciones metábolicas
pueden desembocar en situaciones patológicas. Las patologías infecciosas
serían el resultado de la presencia exógena de PrPSc, es
decir del catalizador o efector o del núcleo. Las patologías
hereditarias ocurrirían por una desestabilización de la estructura de
PrPC o una estabilización de la estructura de PrPSc
favoreciendo la población del estado patológico. Por último, las
enfermedades esporádicas, aunque de etiología desconocida, podrían
surgir por alteraciones metabólicas o bien mutaciones espontáneas que
conlleven la formación de PrPSc. Ambos fenómenos aún
ocurriendo en una única célula podrían desencadenar la formación de
PrPSc y su autopropagación, que se extendería por el sistema
nervioso central().
Todos los enfermos
de vECJ son homocigotos para metionina en el codon 129 del gen PrP. Si
tenemos en cuenta que, aproximadamente, el 40% de la población general
tiene ese genotipo, 10% son homocigotos para valina, y 10% heterocigotos,
no es sorprendente que los primeros casos de vECJ sean homocigotos para
metionina().
La patogenia de las
encefalopatías espongiformes transmisibles ha sido mejor estudiada en el
scrapie. El tracto gastrointestinal y los ganglios linfáticos abdominales
son los primeros órganos infectados, y un año después aparece la
infección en el cerebro. La afectación del tracto gastrointestinal
implica que, el agente infeccioso probablemente infecta las ovejas a través
de la vía oral. En modelos experimentales de scrapie en ratones y cabras,
tras la inoculación subcutánea, el agente patógeno también coloniza
inicialmente en los tejidos linfáticos y bazo, antes de poder ser
detectada en el sistema nervioso central(1).
Incidencia
Encefalopatía
Espongiforme Bovina
En 1987 Wells y
cols describieron cambios espongiformes en el cerebro de una vaca afectada
de EEB(27).
El origen de la epidemia de EEB podría estar relacionado con un cambio en
el proceso de explotación ganadera en 1981, de forma que los priones de
EEB no fueron completamente inactivados antes de alimentar a las vacas con
carne y huesos procedentes de otras vacas infectadas(4).
Hasta el momento, a
nivel mundial, se han declarado 182.507 reses enfermas de EEB (179.441 en
el Reino Unido). La mayor incidencia tuvo lugar en 1992 (37.316 casos a
nivel mundial, de las que 37.056 correspondían al Reino Unido), y desde
entonces la frecuencia global ha disminuido hasta 1.830 casos en el año
2000, por el descenso de casos en el Reino Unido (1.312 casos en el año
2000), aunque en el resto de los países europeos la incidencia ha
aumentado en estos últimos años. Hasta la fecha, se han confirmado 13
casos en España(28).
En Gran Bretaña,
fueron sacrificadas más de 2 millones de reses. En diciembre de 1999, el
Ministerio de Agricultura, Pesa y Alimentación declaró la cabaña británica
"segura" y libre de EEB(4).
La incidencia de
encefalopatía espongiforme en gatos en el Reino Unido también ha
disminuido significativamente desde 1994(26).
Enfermedad
de Creutzfeldt Jacob clásica
La ECJ en su
variante esporádica o no familiar es la enfermedad por priones más
frecuente (85%) en humanos. Las enfermedades por priones en humanos
ocurren en todo el mundo, con una incidencia global de 1 caso por cada
millón de habitantes al año en la enfermedad esporádica y de 1 caso por
cada 10 ó 100 millones de habitantes en la enfermedad familiar. No hay
predominio de sexos(4).
En la ECJ clásica
no se han descrito factores de riesgo no iatrogénicos. Se han descrito
agrupaciones de casos en Israel, Eslovaquia, Chile y Francia, que
corresponden a casos de ECJ familiar con mutaciones en el gen PRNP(4).
Tabla
2. Número total de muertes por ECJ y GSS (incluye casos
definitivos y probables: esporádicos, familiares e iatrogénicos)
(29)
|
|
|
|
|
|
|
|
|
|
|
|
|
Australia
|
Austria
|
Canadá
|
Francia
|
Alemania
|
Italia
|
Holanda
|
Eslovaquia
|
España
|
Suiza
|
Reino
Unido
|
1993
|
21
|
6
|
-
|
55
|
21
|
36
|
12
|
5
|
19
|
10
|
46
|
1994
|
12
|
10
|
2
|
59
|
72
|
38
|
19
|
5
|
17
|
10
|
59
|
1995
|
21
|
10
|
3
|
74
|
87
|
34
|
8
|
6
|
19
|
9
|
44
|
1996
|
27
|
11
|
4
|
88
|
80
|
54
|
16
|
6
|
25
|
10
|
50
|
1997
|
24
|
7
|
15
|
91
|
107
|
60
|
19
|
7
|
28
|
10
|
70
|
1998
|
27
|
8
|
20
|
100
|
111
|
64
|
19
|
6
|
56
|
9
|
71
|
1999
|
19
|
8
|
27
|
96
|
86
|
83
|
19
|
6
|
34
|
8
|
69
|
2000*
|
7
|
3
|
10
|
36
|
43
|
33
|
7
|
3
|
5
|
2
|
22
|
Total
|
158
|
63
|
82
|
599
|
607
|
402
|
119
|
44
|
203
|
68
|
431
|
En España, la cifra de mortalidad de las enfermedades priónicas es
de 0,69 por millón, algo inferior al resto de los países Europeos
(desde 0,97 en Reino Unido hasta 1,37 en Francia), observándose un
discreto aumento en esta frecuencia entre 1993 y 2000 en todos
los países europeos (excepto en Suiza), algo más marcado en el año
1998(29).
El kuru ha
desaparecido, una vez que la población afectada abandonó los hábitos
de canibalismo(4).
Nueva
variante de ECJ
Esta forma
de enfermedad por priones se ha descrito, entre 1995 y enero de 2001,
en 91 pacientes: 88 pacientes británicos, un caso en Irlanda y 2
casos en Francia (4,).
La tabla 3
muestra el número de muertes de casos probables y definitivos en el
Reino Unido, hasta el 28 de Diciembre de 2000(30).
Tabla
3. Muertes por casos probables y definitivos en el Reino Unido
|
|
|
|
|
|
|
|
|
Año
|
ECJ
Esporádico
|
ECJ
Iatrogénico
|
ECJ
Familiar
|
GSS
|
vECJ
confirmados*
|
vECJ
probable aún vivo
|
Total
|
vECJ
muertes esperando resultados autopsia
|
1990
|
28
|
5
|
0
|
0
|
-
|
|
-
|
33
|
1991
|
32
|
1
|
3
|
0
|
-
|
|
-
|
36
|
1992
|
43
|
2
|
5
|
1
|
-
|
|
-
|
51
|
1993
|
38
|
4
|
2
|
2
|
-
|
|
-
|
46
|
1994
|
51
|
1
|
4
|
3
|
-
|
|
-
|
59
|
1995
|
35
|
4
|
2
|
3
|
-
|
|
3
|
47
|
1996
|
40
|
4
|
2
|
4
|
-
|
|
10
|
60
|
1997
|
59
|
6
|
4
|
1
|
-
|
|
10
|
80
|
1998
|
63
|
3
|
4
|
1
|
-
|
|
18
|
89
|
1999
|
61
|
6
|
2
|
0
|
-
|
|
15
|
84
|
2000+
|
38
|
0
|
2
|
0
|
5
|
|
25
|
72
|
Total
|
488
|
36
|
30
|
15
|
5
|
|
81
|
657
|
+
Hasta el 28 de Diciembre de 2000.
Número total de casos definitivos y probables de vECJ =88
*incluyendo 7 muertes probables de vECJ sin conformación neuropatológica.
(Tabla
actualizada el 08/01/01)
La cifra de muertes por la vECJ se mantuvo relativamente constante
hasta que a finales de 1998 Will y cols. describieron una cifra
anormalmente alta de muertes por vECJ. Hasta el 28 de diciembre de
2000, en el Reino Unido, se detectaron 81 muertes por vECJ y otros 7
casos probables (30,31).
En 1998, se
describió por primera vez en España un caso de vECJ en un paciente
de 27 años, inglés, procedente de Souhampton(32),
pero hasta el momento no se ha comunicado ningún caso en residentes
españoles.
En el Reino
Unido entre 1993 y 2000 (7,5 años de estudio) se ha descrito un total
de 501 casos de ECJ, incluyendo la vECJ, 34 casos de ECJ familiar (25
con mutación PRNP probada), y 28 casos iatrogénicos. En ese mismo
periodo, en España (donde aún no se ha descrito la vECJ) se observó
un total de 203 casos de ECJ, 11 casos de ECJ familiar (10 con mutación
PRNP probada) y dos casos iatrogénicos (por implantes de
duramadre)(29).
Puesto que la
vECJ se caracteriza por aparecer en pacientes más jóvenes que la
forma clásica de la enfermedad, es interesante conocer el número de
casos de ECJ clásica que se han observado antes de los 50 años
(tabla 4):
Tabla
4. Número de ECJ con edad < 50 (1996- 30/6/00)
y Número de casos de vECJ (muertes hasta 30/6/00)
|
|
|
PAÍS
|
NO.
CASOS EDAD <50 DESDE 1996
|
No.
CASOS vECJ
|
Australia
|
37**
|
0
|
Austria
|
18*
|
0
|
Canadá
|
9
|
0
|
Francia
|
317
|
2
|
Alemania
|
75*
|
0
|
Italia
|
92*,
27º, 64**
|
0
|
Holanda
|
9
|
0
|
Eslovaquia
|
4
|
0
|
España
|
7
|
0
|
Suiza
|
9
|
0
|
Reino
Unido
|
198**
|
70
|
La
tabla 5 muestra la distribución por edades de 52 casos de vECJ(33).
Tabla
5. Distribución vECJ por edades
|
|
Edad
Muerte
|
Número
de casos
|
0
- 9
10 - 19
20 - 29
30 - 39
40 - 49
50 - 59
59+
Total
|
0
8
23
14
3
4
0
52
|
Características
clínicas de enfermedades priónicas en humanos
Enfermedad
de Creutzfeldt-Jakob esporádica (ECJ)
La tétrada
clásica de la ECJ es: confusión y pérdida de memoria que progresan
rápidamente a demencia, junto a ataxia, mioclonia, y
electroencefalograma anormal, y aparece en un 60% de los casos. Otros
signos y síntomas neurológicos descritos son: debilidad focal o
generalizada, neuropatía dolorosa, movimientos coreiformes,
alucinaciones, ceguera cortical, trastornos del lenguaje,
oftalmoplegia supranuclear, y síndrome de manos ajenas. En un 25% de
los casos, los síntomas iniciales son cansancio, dolor de cabeza,
trastornos del sueño, vértigo y alteraciones del comportamiento(4).
Una vez
manifiesta la enfermedad, el ritmo de progresión es rápido, y el
paciente termina postrado en cama, sin comunicarse, aunque puede
mantener un ciclo vigilia-sueño y movimientos oculares espontáneos.
La duración media de la enfermedad es de 4 a 5 meses, y la muerte
suele suceder por complicaciones respiratorias o sepsis relacionada
con la incontinencia(4).
Durante el
curso de la enfermedad, el EEG muestra paroxismos pseudo-peródicos de
ondas trifásicas o picudas de 0,5 a 2,0 Hz, sobre un fondo lento. Sin
embargo, un 30% de los enfermos pueden no mostrar éste EEG típico,
sobre todo al principio o en los estadios más avanzados de la
enfermedad. Por ello, se recomienda EEG semanales para poder detectar
el patrón característico(4).
El examen del líquido
cefalorraquídeo, generalmente es negativo y sólo muestra una
incremento ligero de proteínas. La detección de la proteína 14-3-3
en líquido cefalorraquídeo permite excluir otros tipo de demencia(34).
La sensibilidad del inmunoensayo (Western Blot) para la detección de
la proteína 14-3-3 es de un 97%, aunque su sensibilidad es de un 87%
pues existen algunos falsos positivos, sobre todo en casos de
accidente cerebrovascular o meningoencefalitis(35).
La tomografía
axial computadorizada (TAC) y la resonancia nuclear magnética (RNM)
son útiles para excluir otras causas de enfermedad neurológica
subaguda(4).
Actualmente la resonancia nuclear magnética (RNM) es capaz de
diagnosticar alteraciones altamente específicas, como señales hiper-intensas
en núcleos de la base, en sólo el 67% de los enfermos de Creutzfeldt-Jakob(36),
aunque algunas series describen una sensibilidad de un 100%(37).
En las
enfermedades por priones, excepto en el insomnio fatal, la tomografía
de emisión de positrones (PET) sólo muestra un hipometabolismo
cortical inespecífico(4).
Conviene
recordar que algo menos de un 20% de los casos tienen una presentación
clínica atípica, como comienzo brusco (variante Heidenhain), duración
prolongada, o EEG sin cambios característicos. Algunos de estos casos
se acompañan de cambios anatomopatológicos peculiares, como la
afectación exclusiva del lóbulo occipital. Algunos casos, que se
presentan con un predominio de ataxia cerebelosa, son homocigotos para
Val en codon 129, y se describe afectación predominante del
tronco cerebral y cerebelo. En Japón se ha descrito una forma pan-encefalopática
de ECJ, con extensa afectación de sustancia blanca, con hallazgos en
resonancia nuclear magnética, similares a la leucodistrofia(4).
Diagnóstico diferencial
Dada la
gran variabilidad de síntomas de presentación en la ECJ, los diagnósticos
diferenciales a considerar incluyen: enfermedad de Alzheimer,
demencias frontotemporales, demencia con cuerpos de Lewi,
lipofucsinosis ceroide (enfermedad de Kufs en el adulto), enfermedad
de Huntington, ataxias espinocerebelosas, esclerosis lateral amiotrófica
y esclerosis múltiple, entre otras(4).
El rápido
progreso de la ECJ esporádica puede hacer pensar en vasculitis del
sistema nervioso central, aunque la RNM y la arteriografía cerebral
permiten descartar este diagnóstico. Las causas metabólicas que
pueden similar enfermedades por priones son las intoxicaciones por
bismuto, mercurio y litio, y la tiroiditis autoinmune. Este último
trastorno puede presentar episodios intermitentes de confusión y
ataxia cerebelosa e incluso mostrar las descargas periódicas en EEG.
Los niveles de anticuerpos anti-peroxidasa tiroidea y anti-tiroglobulina
está elevados, incluso cuando el nivel de TSH es normal. El
tratamiento con conticoesteroides puede aliviar completamente los síntomas(4).
El análisis
genético del gen PRNP puede ayudar, exista o no una historia familiar
de ECJ, pues la historia familiar puede verse oscurecida por el
comienzo tardío y a menudo variable, en el comienzo de los síntomas
en estas enfermedades. La detección de una mutación del gen PRNP
sugeriría el diagnóstico de enfermedad por priones, pero debemos
recordar que la encefalitis herpética, la vasculitis, la intoxicación
medicamentosa, y otras enfermedades potencialmente tratables, también
pueden darse en pacientes con mutación en PRNP(4).
Ante la falta
de pruebas de laboratorio definitivas (tabla 6), en los pacientes jóvenes
(30 a 50 años) se plantea la realización de una biopsia cerebral
para llegar a un diagnóstico definitivo(4).
La última
prueba de diagnostico definitivo sería la transmisión de la
enfermedad a líneas especiales de ratones que llevan el gen humano
PrP (ratones transgénicos), que son muy susceptibles a enfermedad por
priones humanos. Sin embargo, el periodo de incubación es largo (200
días)(4).
Tabla
6 Criterios diagnósticos clínicos de la OMS para el
diagnóstico de ECJ ()
|
Diagnóstico
definitivo de ECJ
Neuropatología
ECJ Probable
(1) Demencia progresiva
(2) EEG típico (complejos periódicos trifásicos
generalizados de 1 Hz)
(3) Al menos dos de los siguientes:
• Mioclonia
• Trastornos
de visión o cerebelosos
• Disfunción piramidal/extrapiramidal
• Mutismo
aquinético
ECJ Posible
(1) Los mismos criterios descritos arriba
(2) EEG no disponible o no es típico
(3) Duración de menos de 2 años.
|
Anatomía
Patológica de la ECJ
En todos
los casos de ECJ, por definición, se identifica vacuolización de
la sustancia gris (cambio espongiforme o espongiosis) (figura 3).
Las vacuolas observadas en el microscopio óptico son debidas a
hinchazón focal de axones y terminaciones neuronales dendríticas
asociado a la pérdida de organelas sinápticas y la acumulación de
membranas anormales. Aunque la espongiosis predomina en la sustancia
gris, puede ser también observada en el neuropilo de la sustancia
blanca subcortical o profunda. Dentro del encéfalo, la distribución
típica es en el neocórtex cerebral, subiculum del hipocampo,
caudado, putamen, tálamo, y la capa molecular de la corteza
cerebelosa(4).
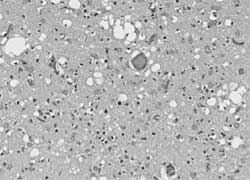
Figura 3.
Espongiosis en sustancia gris del cerebro en ECJ esporádica(30)
Las vacuolas deben demostrarse también dentro de los citoplasmas de
las neuronas. No se identifica respuesta inflamatoria, pero sí se
aprecia gliosis reactiva().
La PrP
resistente a proteasa puede ser detectada fácilmente en el cerebro
de estos pacientes mediante inmunohistoquímica(4).
Forma
familiar de ECJ
Dentro de
la forma familiar de ECJ se incluyen los casos con una mutación del
gen PRNP heredada de forma dominante, en los que los cambios
anatomopatológicos demuestran cambios espongiformes sin placas tipo
GSS(4).
Aunque las
formas familiares de ECJ tienden a presentar una fenotipo clínico y
neuropatológico similar a la ECJ esporádica, se aprecian algunas
diferencias. Los pacientes con ECJ tienen una edad media de comienzo
de síntomas unos 12 más precoz y la duración de la
enfermedad es superior a la forma esporádica en unos 18 meses. Por
otra parte, algunas mutaciones del gen PRNP dan lugar a síndromes
clínicos distintos al cuadro clínico típico de la ECJ esporádica(4).
No se conoce
la utilidad de la proteína 14-3-3 en LCR en el diagnóstico de ECJ
familiar o en la enfermedad por priones atípica(4).
Forma
Iatrogénica de ECJ
La ECJ se
ha descrito en más de 100 pacientes que recibieron hormona del
crecimiento humana (hGH) procedentes de glándulas pituirarias
humanas. El riesgo de enfermedad se correlacionada con la duración
del tratamiento con hGH.
A diferencia
de la ECJ clásica, estos pacientes presentan más a menudo ataxia
cerebelosa en vez de disfunción cortical superior, y el EEG muestra
un patrón de ondas lentas en vez de las descargas periódicas trifásicas
observadas en la ECJ esporádica.
Aunque el uso
de hormona del crecimiento recombinante empezó en 1985, aún se
observan casos de ECJ iatrogénica, debido al prolongado periodo de
incubación de la enfermedad, de hasta 20 años(4).
También se
ha descrito pacientes con ECJ tras implantes de duramadre,
trasplantes de córnea, exposición a electrodos para la localización
de focos de epilepsia, y la gonadotropina de pituitaria humana(4).
El periodo de
incubación de la ECJ iatrogénica es de 15 meses a 20 años(1).
Aunque los
estudios experimentales indican que la sangre puede ser infecciosa
en algún momento durante el curso de la enfermedad, no hay
evidencias que indiquen la existencia de un riesgo significativo de
transmisión de enfermedad por priones a través de la sangre o de
derivados sanguíneos. Además, los estudios epidemiológicos no
apoyan la existencia de riesgo significativo de transmisión de ECJ
a través de transfusión sanguínea(4).
Enfermedad
de Gerstmann-Straussler-Scheinker (GSS)
Esta
enfermedad fue descrita en una familia alemana en la que se observó
la aparición antes de los 50 años, de ataxia y disartria, seguida
de grados variables de síntomas piramidales y extrapiramidales, y
la aparición tardía de demencia. El EEG no muestra los cambios típicos
observados en la ECJ. La enfermedad tiene una duración de 2 a 10 años.
El estudio neuropatológico muestra depósito de placas de amiloide
en ciertas área o por toda la corteza cerebral, que reaccionan con
los anticuerpos anti-PrP humano. El grado de espongiosis es mínimo(4).
Insomnio
fatal (IF)
Se
describen casos familiares (IFF) y esporádicos de esta enfermedad.
La edad de comienzo oscila entre los 25 y los 61 años (media: 45 años)
y el tiempo medio de duración de la enfermedad, hasta el
fallecimiento, es de 1 a 2 años. La presentación más típica es
la aparición de insomnio intratable, a veces durante semanas o
meses. El insomnio es seguido de disautonomía, ataxia, y síntomas
piramidales y extrapiramidales variables. Las alteraciones disautonómicas
pueden dar lugar a alteraciones en la tensión sanguínea,
frecuencia cardiaca, temperatura, ritmo respiratorio, y secreciones
corporales. El EEG muestra enlentecimiento difuso en vez de
descargas periódicas. La PET muestra una reducción de la actividad
metabólica o del flujo sanguíneo en el tálamo(4).
Los cambios
neuropatológicos del IF incluyen pérdida neuronal y astrogliosis
en el tálamo y en las olivas inferiores, y, en menor grado, en el
cerebelo. La vacuolización es mínima o está ausente en los casos
típicos. La PrP proteasa resistente es detectable, pero está
presente en mínimas cantidades y sólo en el tálamo y en el
lóbulo temporal(4).
El insomnio
fatal familiar probablemente es la misma enfermedad que la llamada
"demencia puramente talámica", pues estos enfermos tienen
la misma mutación D178N observada en IFF(4).
El prión PrPSc
encontrado en el cerebro de los pacientes de IFF es distinto al de
pacientes con ECJ familiar. Tras digestión limitada con proteasa y
electroforesis, el PrP resistente a proteasa de IFF migra a 19 kDa y
el de la ECJ familiar lo hace a 21 kDa(4).
La aparición
de casos de IF esporádicos, además del IFF, apoya la idea de que
la conformación de la proteína PrP, y no tanto la secuencia de
aminoácidos, es el determinante principal del fenotipo de la
enfermedad(4).
Variante
de la ECJ
La clínica
de la vECJ es notablemente distinta a la ECJ esporádica, pues el
comienzo se asocia con síntoma psiquiátricos, aparece sobre todo
en gente joven (media: 27 años, rango: 16 a 48 años), con un curso
algo prolongado de unos 16 meses (rango: 9 a 38 meses) y no se
asocia con complejos periódicos en el EEG(4).
La RNM
muestra aumento de intensidad en putámen en imágenes T2(4).
El cerebro
muestra vacuolización difusa y presencia de placas características
de núcleo denso que contienen PrP, rodeadas de un halo de cambio
espongiforme, denominadas "placas floridas" (figura 4)(4).
En el LCR de
enfermos de vECJ no se detecta proteína 14-3-3(4).
La separación de este cuadro como una entidad clínica separada de
la ECJ clásica (tablas 7 y 8) viene apoyada por el hallazgo
de un PrPSc en el cerebro de los pacientes de vECJ
predominantemente bi-glucosilada, con distinta movilidad electroforética
a la de la ECJ esporádica, la cual es predominantemente
monoglucosilada. El mismo tipo de proteína se observa tras la
transmisión experimental de EEB a varios animales, lo que apoya la
idea de que la vECJ es consecuencia de una infección de humanos por
EEB(4).
Tabla
7. Diferencias entre la ECJ clásica y la vECJ (4,26)
|
|
|
|
| |
ECJ
clásica esporádica
|
ECJ
clásica familiar
|
vECJ
|
Frecuencia
|
75-85%
de casos
|
5-10%
de casos
|
13,3%
de casos
|
Edad
de inicio
|
Mayor
de 60 años (17-83), rara < 40 años
|
Variable.
Menor de 60 años (20 - 80)
|
16
a 52 años (Adolescentes y adultos jóvenes)
|
Duración
de la enfermedad
|
Corta:
4 a 6 meses
|
Larga:
1 a 5 años
|
Larga:
14 meses
|
Primeros
síntomas
|
Neurológicos:
Demencia, ataxia, mioclonias
|
Neurológicos:
Demencia, ataxia, mioclonias
|
Psiquiátricos,
demencia tardía
|
EEG
|
Característico
|
Característico
|
No
Característico
|
Anatomía
Patológica
|
Espongiosis
difusa y gliosis de la sustancia gris. Pocas o ninguna placa
amiloide PrP
|
Espogiosis
difusa y gliosis de la sustancia gris
|
Placas
"floridas" (placas PrP con dentro denso rodeadas de
un halo de cambio vacuolar), con extensa afectación
cerebelosa, y espongiosis difusa
|
Proteína
del prión
|
Monoglucosilada
|
Migra
a 21 kDa
|
Altas
concentraciones de forma bi-glucosilada
|
Relación
con otras especies
|
|
|
El
agente causal es idéntico a la EEB y EEF (felinos)
|
Gen
PrP
|
Factores
de riesgo: Polimorfismo u homocigosidad en el codon 129
|
Mutaciones
puntuales, inserciones / delecciones
|
Homozigotos
para metionina en codon 129
|
Tabla
8. Otras enfermedades por priones en humanos(4)
|
|
|
|
| |
Kuru
|
GSS
|
Insomnio
Familiar
|
Edad
de inicio
|
40
años (29-60)
|
Variable.
Menor de 60 años (20 - 80)
|
45
años
|
Duración
de la enfermedad
|
Corta:
3 mesas a 1 año
|
Larga:
2 a 6 años
|
Larga:
1 a 2 años
|
Primeros
síntomas
|
Neurológicos:
Ataxia y demencia.
|
Neurológicos:
Ataxia, demencia tardía
|
Neurológicos:
Insomnio, disautonomía,
ataxia, demencia
|
EEG
|
|
|
|
Anatomía
Patológica
|
Afecta
sobre todo al cerebelo: Cambio espongiforme difuso, pérdida
neuronal, y depósitos amiloides de centros densos
("placas de kuru")
|
Numerosas
placas amiloides PrP-positivas, gliosis y haces
neurofibrilares, Espongiosis mínima.
|
Gliosis
focal talámica y olivar, sustitución neuronal. Mínima
espongiosis
|
Tratamiento
de las enfermedades por priones
Actualmente, no hay ningún tratamiento farmacológico disponible
para las enfermedades por priones, por lo que sólo pueden tomarse
medidas sintomáticas. Experimentalmente, los compuestos
polisulfatados, como el pentosán polisulfato, suramina, y el heparán
sulfato, parecen retrasar la aparición de scrapie en animales, y
afecta a la generación de PrPSc en cultivos celulares.
Sin embargo, los efectos son mínimos y los compuestos deben ser
administrados antes de la inoculación del animal para tener efecto(4).
El consejo
genético es un aspecto fundamental en el manejo de la enfermedad
por prión familiar. Aunque es discutible si las pruebas genéticas
deben realizarse en los miembros asintomáticos de la familia, la
administración Norteamericana ha prohibido la donación de sangre a
los familiares de pacientes con enfermedades por priones, a menos
que el análisis genético del donante potencial demuestre una
secuencia normal del gen PRNP(4).
Genética
de las encefalopatías espongiformes transmisibles
Se estima que cerca del 15% de los casos de enfermedades por priones
en humanos se heredan de forma autosómica dominante(4).
Todos los
hallazgos indican que el aminoácido situado en la posición 129
tiene una función esencial en las interacciones PrPSc-PrPC
, en la conformación de la PrP y en el tipo de enfermedad que
se desarrollará.
En la ECJ clásica
se han descrito 7 mutaciones puntuales diferentes (por ejemplo, Val
à Ile) y 5 tipos de delecciones/inserciones distintas del gen PRNP(4).
En GSS se han descrito 6
mutaciones puntuales diferentes (por ejemplo, Ala à Val) y 2 tipos
de delecciones/inserciones distintas del gen PRNP. En el IF se ha
descrito la mutación puntual Asp àAsn del mismo gen(4).
La existencia
de polimorfismo en el codon 129 del gen PRNP parece que actúa como
un factor de susceptibilidad genética. En esa posición se codifica
Metionina (Met) o Valina (Val). En la población caucásica, la
distribución de genotipos es: 37% Met/Met, 51% Met/Val, y 12%
Val/Val, es decir, un 50% aproximadamente es homocigoto para Met o
Val y un 50% heterocigoto. En algunos estudios sobre la ECJ clásica
esporádica y ECJ iatrogénica por hormona del crecimiento
contaminada, la prevalencia de homocigotos para Met o Val en el
codon 129 es de un 90%, lo que sugiere que la homocigosidad en el
codon 129 es un factor de riesgo para esta enfermedad por priones(4).
Por otro lado, en todos los casos conocidos de vECJ los genotipos de
los enfermos son homocigotos para Met(31).
El genotipo
en el codon 129 también afecta al curso de la enfermedad. De esta
forma, los enfermos de ECJ clásica esporádica homocigotos para Met
se asocian generalmente a una enfermedad demenciante rápidamente
progresiva, mientras que el genotipo Val/Val se asocia a un curso más
prolongado con aparición de ataxia(4).
Por otro
lado, en algunas formas familiares de la enfermedad, la
homocigosidad en el codon 129 se asocia con una menor edad de
aparición y un curso más agresivo de la enfermedad(4).
La frecuencia de distribución de genotipos varía con las razas, así,
mientras que entre los caucásicos la proporción es Met (0.66):Val
(0.34), las frecuencias alélicas en descritas en Japón son Met(0.96):
Val(0.04). La incidencia de ECJ en Japón no es mayor que en el
resto del mundo, por lo que la homocigosidad en el codon 129 no
predice un mayor riesgo de enfermedad, pero predispone a un
individuo a padecer el fenotipo clínico de ECJ si se suman otros
factores de riesgo esenciales(4).
Los
portadores de mutaciones puntuales (como PRNP-E200K) muestran
un mayor riesgo de desarrollar la enfermedad conforme avanza la edad
de los pacientes(4).
La mutación
más frecuente en la ECJ en todo el mundo es la sustitución de
glutamato (E) por lisina (K) en el codon 200: E200K (Glu à Lys).
También son frecuentemente observadas las siguientes mutaciones:
D178N (Asp à Asn en ECJ, ó Asp àAsn en IF), y la inserción de
144-bp(4).
La mutación más frecuente en GSS es P102L (Pro àLeu)(4).
En todos los pacientes de vECJ el codon polimórfico 129 es
homocigoto para Met, sugiriendo que la homocigosidad para Met
incrementa la susceptibilidad a vECJ. Otra posibilidad es que los
pacientes con genotipo homocigoto para Val no desarrollan la patología
característica descrita en la vECJ(4).
Transmisión
de EET en humanos.
La
naturaleza transmisible de las enfermedades por priones fue
demostrada por primera vez experimentalmente en 1936, cuando Cuillé
y Chelle transmitieron scrapie a una cabra sana mediante la
administración intraocular de médula espinal infectada de scrapie().
Treinta años más tarde, Gajdusek y cols (1966) consiguieron
transmitir kuru a chimpancés().
En 1968 Gibbs
y cols. transmitieron la ECJ esporádica a chimpancés().
En la década de 1960, Carleton Gajdusek confirmó que la enfermedad
de Creutzfeldt-Jakob era transmisible(5). A pesar de
haber transcurrido más de tres décadas desde que se comenzó la
investigación de la transmisión de enfermedades por priones, la
información hoy día disponible se puede definir como
fragmentada y e incoherente ().
Incluso antes
de conocerse la relación entre la vECJ y el "mal de las vacas
locas", se sabía que existía una enfermedad de Creutzfeldt-Jakob
(llamada iatrogénica) que podía ser transmitida por la
administración de la hormona de crecimiento humana y la hormona
gonadotropina procedentes de cadáveres, o mediante trasplantes de córnea
o de duramadre(11).
En las enfermedades priónicas en humanos, la mayoría de los casos
son esporádicos (son casos generalmente de Enfermedad de Creutzfeld-Jakob),
pero también es importante la transmisión familiar o genética (en
la Enfermedad de Creutzfeld-Jakob, la enfermedad de Gerstmann-Straussler-Scheinker,
el Insomnio Familiar Fatal y la Demencia Talámica). Otras vías de
transmisión son la iatrogénica (descrita anteriormente) y, en la
enfermedad del kuru por ingesta, en relación con rituales de
canibalismo en tribus Fore en Papua, Nueva Guinea(6).
Ciertas circunstancias en
el Reino Unido causaron la aparición y propagación de la EEB en el
ganado, entre estas circunstancias se encuentran, por una parte, el
empleo frecuente de carne y huesos en piensos que procedían de
ovejas infectadas de scrapie y por otra, la adopción de un nuevo
tipo de procesamiento que no reducía la cantidad de priones
infecciosos tras la elaboración del pienso().
La EEB también ha sido
transmitida a rumiantes salvajes en zoológicos del Reino Unido, que
fueron infectados al comer los mismos piensos concentrados de carne
y huesos contaminados, responsables de la enfermedad en el ganado
bovino. También se ha descrito la encefalopatía espongiforme en más
de 80 gatos y en algunos felinos de zoológicos británicos(26).
Hoy en día
los estudios se están centrando en probar la ingesta como un factor
de riesgo real(43).
Experimentalmente, se ha confirmado que la encefalopatía
espongiforme bovina puede ser transmitida por vía oral e
intracerebral a diferentes especies animales (incluyendo primates)
con mayor o menor facilidad, aunque, sobre todo, es posible pasarla
por vía oral de una vaca a otra con apenas la ingestión de pequeñas
cantidades de cerebro(). El patrón de la EEB se mantiene
a pesar de su paso a un número variable de animales intermedios.
Esta firma del prión de la EEB también había sido identificada en
animales exóticos y gatos domésticos infectados con la EEB. La
firma de la EEB presentaba una gran homogeneidad en su
comportamiento y una gran ubicuidad para infectar diversas especies
de animales, este hecho no se había visto antes con otros agentes
priónicos desde 1963(45).
La transmisión de ECJ
esporádica a primates humanos se consigue en un 85% de los casos.
La inoculación a ratones transgénicos que expresan la PrP humana
es aún más eficaz, consiguiéndose en casi un 100% de los
casos. Esta mejor transmisión en ratones transgénicos que expresan
la PrPC humana demuestra la importancia de la homología
de secuencia entre PrPSc y PrPC y
ayuda a explicar la dificultad en la transmisión entre especies con
secuencia PrP distintas, un hecho conocido como "barrera de las
especies". Sin embargo, la aparición de la vECJ y la transmisión
de priones humanos a ratones transgénicos que expresan PrPC
bovino hacen pensar que la secuencia de PrP no es el único factor
importante para la transmisión de la enfermedad. El factor
determinante puede ser la capacidad de PrPC para adoptar
la conformación de PrPSc, la cual debe estar influida
tanto por la secuencia de la proteína como por otros factores (ej.
proteínas carabina)(4).
El grado de
transmisión de GSS a primates no humanos es bajo (40%), comparado
con la ECJ. Además, los intentos de transmitir GSS a roedores no
han tenido éxito. La escasa capacidad de transmisión de esta
enfermedad podría estar relacionada con la escasez relativa de PrP
proteasa resistente en el cerebro de los pacientes(4).
Hasta la
fecha, la Unidad de Vigilancia de la ECJ del Reino Unido no ha
detectado ningún caso de vECJ consecuencia de procedimientos quirúrgicos
o médicos(1).
Trasmisión
por derivados de la sangre
La
preocupación actual entre los científicos se centra en la posible
transmisión de la enfermedad de Creutzfeldt-Jakob a través de
sangre o derivados de la sangre, aunque aún no hay datos firmes
epidemiológicos que apoyen este modo de transmisión(4),
sí hay datos de estudios experimentales en laboratorios que
conforman que la enfermedad Creutzfeldt-Jakob clásica (no la
variante) puede ser transmitida a través de componentes del plasma
sanguíneo, aunque su grado de infectividad es mucho menor que
en los tejidos cerebrales(46).
Algunas
directivas de la Comunidad Europea (como las del Grupo de
Biotecnología del Comité de Productos Médicos Patentados) regulan
la comercialización de productos derivados de la sangre (incluyendo
por ejemplo plasma, o excipientes de medicamentos). De momento sólo
incluyen directamente la exclusión de material procedente de
pacientes con enfermedad de Creutzfeldt-Jakob clásica (ya sea esporádica,
familiar o iatrogénica), mientras que en el caso de donantes que
sufren de la vECJ, desde febrero de 1998, se aconseja descartar este
material por precaución, y desde enero de 1999 se recomienda la
realización de exámenes de detección de encefalopatías
espongiformes transmisibles, en los derivados de la sangre(47),
a pesar que estudios experimentales recientes indican que los
procesos realizados durante la producción de productos derivados
del plasma parecen eliminar agentes priónicos de forma
significativa (48).
Es posible
transmitir la EEB a ovejas mediante la transfusión de sangre entera
procedente de otras ovejas asintomáticas alimentadas con reses
infectadas de EEB, lo que demuestra que la EEB puede ser transmitida
entre individuos de la misma especie a través de transfusión de
sangre completa. Sin embargo, aún no hay evidencias directas de
transmisión de vECJ a través de transfusión sanguínea en humanos(49).
La detección
de proteína priónica anormal en las amígdalas de individuos
infectados por el vECJ ha levantado las sospechas de que la sangre
también puede ser una vía de transmisión en la vECJ, incluso más
importante en la enfermedad de Creutzfeldt-Jakob clásica, pero este
riego aún está por definir(48).
El
Departamento de Salud del Reino Unido ha anunciado que no hay
evidencia de transmisión de la vECJ a humanos a través de
transfusiones sanguíneas. El riesgo es sólo teórico, pues la
sangre completa ya no es utilizada para transfusiones en ese país,
puesto que los leucocitos son previamente extraídos mediante
leucodeplección. Además, en ese país, el plasma para la elaboración
de productos sanguíneos es importado de países con baja incidencia
de EEB y sin vECJ(49).
Para evitar
la transmisión de este tipo de enfermedades a través de
instrumental médico, dada la alta resistencia de este agente
infeccioso a muchos métodos de descontaminación y esterilización,
se aconseja la utilización de soluciones esterilizantes, como las
basadas en ácido peracético para inactivar los priones(50).
Aunque en
Estados Unidos no se han descrito casos de EEB en animales ni vECJ
en humanos, desde Agosto de 1999, el Departamento de Salud de este
país prohibieron la donación de sangre de personas que hubiesen
residido en Gran Bretaña 6 meses o más entre 1980 y 1996. Estas
prohibición podría extenderse a todos los países de Europa
Occidental. Se estima que esta medida reduciría el número de
donantes de sangre un 5 %(51).
Medidas
a tomar
En Julio de 1988, en el Reino Unido se prohibieron los piensos con
proteínas de rumiantes destinados a rumiantes. Esta prohibición
logró una disminución de nuevas infecciones(26). En
1989, la Unión Europea dispuso algunas limitaciones para la venta
de ganado vacuno vivo procedente del Reino Unido, y en 1990 se
consideró la EEB de declaración obligatoria().
Tras la detección de
encefalopatía espongiforme en gatos, en 1990 se prohibió la
utilización productos bovinos peligrosos en la industria de comidas
para animales domésticos. En 1996, se amplió la prohibición a las
proteínas de mamíferos en la alimentación de todas las especies
animales en las granjas(26).
Las medidas
adoptadas por la Unión Europea están resumidas en un reciente
documento disponible en Internet(52).
Una vez
reconocido el riesgo de transmisión de EEB al ser humano, la
principal medida adoptada se adoptó en Noviembre de 1989 al
prohibir el uso de ciertos tejidos de bovinos en la comida para
humanos, sobre todo aquellos en los que se detectó una mayor
concentración del agente infeccioso, como el cerebro, la médula
espinal, las amígdalas, bazo, timo, e intestinos de reses mayores
de 6 meses de edad. Esta prohibición se ha extendido, desde 1996 a
toda la cabeza, excluyendo la lengua. Por otra parte, puesto que la
médula espinal puede no ser totalmente extirpada en algunos
animales, el gobierno británico prohibió también la columna
vertebral en diciembre de 1995 en carne procesada mecánicamente. En
marzo de 1996, este gobierno decide que sólo las reses menores de
30 meses son aptas para el consumo humano. De hecho, sólo se han
descrito 265 casos en reses menores de 30 meses. Al identificarse el
agente infeccioso en los ganglios de las raíces dorsales durante el
periodo de incubación, en diciembre de 1997 el gobierno británico
decide prohibir la carne con hueso, incluso en animales menores de
30 meses. En marzo de 1996, la Unión Europea prohibió la
venta de carne de vaca y productos bovinos procedentes del Reino
Unido(26).
En el momento actual, se
han tomado diversas medidas por el Gobierno Británico acerca de la
depleción de leucocitos de la sangre, así como de la fabricación
de derivados plasmáticos con plasma de fuera del Reino Unido, ante
la simple posibilidad hipotética de transmisión de ECJ por
derivados hemáticos. Recientemente, también se han publicado datos
sobre la posible persistencia de los priones en animales
aparentemente sanos, que podrían actuar como reservorios y
perpetuar la cadena infectiva(45).
Además de las cuestiones
meramente científicas, otra consecuencias de esta crisis ha sido la
revisión de los patrones de conducta más adecuados en el uso y
tratamiento de la información científica para su difusión pública
a la población, tanto por los medios de comunicación como por los
estamentos públicos responsables de la misma. La necesidad de
objetividad y claridad absoluta en la información facilitada por
los responsables oficiales, así como en la necesidad de disponer de
comités de evaluación integrados por personal experto externo y
objetivo, que no pueda presentar conflictos de intereses, parece una
necesidad cada vez más clara(45).
La aparición de los nuevos métodos diagnósticos y el incremento
de la atención de los clínicos ha modificado las condiciones para
el diagnóstico de la ECJ clásica. Los criterios diagnósticos clásicos
fueron diseñados para clasificar casos fallecidos. El desarrollo de
la genética ha permitido ampliar el diagnóstico en casos
portadores de mutaciones con síntomas poco específicos y
caracterizar (polimorfismos del codon 129) grupos de susceptibilidad
demostrada para los agentes transmitidos por iatrogenia y de posible
especial susceptibilidad ante la exposición al agente de la EEB.
Las recientes adopciones de nuevos criterios para clasificación en
categorías diagnósticas por la OMS y el grupo europeo de
vigilancia y estudio de la ECJ suponen la inclusión en los contajes
de incidencia a partir del 1998 de los casos "definitivos o
ciertos" y los "probables," considerando como casos
"probables" los casos "posibles" según Collins
y Masters con positividad al test de la 14-3-3. La introducción del
test en la practica clínica española en el 1997 supondrá un
impacto considerable en las incidencias observadas. La posibilidad
de inclusión por este mecanismo de falsos ECJ 14-3-3 positivos sin
comprobación post-mortem, por la caída de la proporción de
sospechosos con necropsia deberá ser considerada().
La inoculación
de extractos de cerebro, médula espinal, ojo, pulmón, hígado, riñón,
bazo, ganglio linfático y LCR de pacientes con ECJ puede infectar a
primates. Sin embargo, las secreciones, heces y orina no se ha
demostrado que transmitan la infección. Por ello, ante un enfermo
de ECJ sólo son necesarias medidas preventivas habituales, dado que
el riesgo de transmisión a otros miembros de la familia o
cuidadores es bajo. No es necesario el aislamiento completo. Sin
embargo, deben tomarse precauciones especiales al extraer LCR o al
manipular tejidos obtenidos en la autopsia(1).
Es esencial adoptar
medidas de protección para el personal que manipula muestras de
tejidos infectados por priones para poder realizar de forma segura
los estudios neuropatológicos necesarios.
En los laboratorios de anatomía patológica, conviene recordar que
al realizar la autopsia de un caso probable de ECJ, deben congelarse
muestras del cerebro, generalmente del lóbulo frontal y cerebelo,
para poder efectuar un tipaje de proteína de prión y secuenciación
del gen de la proteína del prión. Las muestras de los casos de ECJ
deben ser manipuladas en campanas de aislamiento (clase 1). Los
bloques de tejido que vayan a ser procesados deben ser tratados
utilizando agente que inactive el prión, como la inmersión en ácido
fórmico puro durante al menos una hora(). Los
instrumentos deben ser descontaminados sumergiéndolos en una solución
>1N de hidróxido sódico durante al menos 1 hora y luego
sometiéndolos a autoclave a 134º C durante al menos 1 hora.
Deberán utilizarse instrumentos desechables e incinerar los
materiales contaminados siempre que sea posible(1).
Algunas vacunas están
fabricadas con sustancias procedentes de bovinos de países en los
que existe la EEB. Sin embargo, el riesgo de transmisión de la vECJ
por esta vía se considera remoto y los beneficios de la vacunación
sobrepasan a los remotos riesgos de vECJ. Hasta la fecha no existen
evidencias de casos de vECJ relacionados con el uso de vacunas. Sin
embargo, se recomienda la sustitución paulatina de esas vacunas por
otras donde los productos bovinos procedan de países sin riesgo de
EEB().
En EE.UU., se
estima que un cuarto de las fábricas de piensos no cumplen las
regulaciones dispuestas para prevenir la introducción y diseminación
de la EEB en ese país, al incluir en esos piensos cerebro y médula
espinal().
Se debe
pensar en la nueva variante de ECJ ante un paciente joven que
desarrolle un cuadro de depresión, al que se asocien alteraciones
de la marcha, signos piramidales, extrapiramidales, mutismo acinético
o mioclonias, aunque ocurra en un país donde no se haya descrito aún
la enfermedad(32).
Perspectivas
de futuro
A la vez que se aclaran los mecanismos íntimos de transmisión de
estas enfermedades priónicas, pronto asistiremos a técnicas de
diagnóstico más precoces, tanto de mediante exámenes en líquidos
corporales, como técnicas radiológicas. Estas técnicas
complementan a la electroencefalografía (EEG), que hoy en día es
un importante elemento de ayuda en el diagnóstico de probabilidad
de la enfermedad de Creutzfeldt-Jakob(37).
Desconocemos
si en un futuro más o menos próximo existirá una epidemia de vECJ,
pero conviene destacar que la incidencia de nuevos casos en el Reino
Unido ha permanecido baja durante estos años(4).
Sir John
Pattison, miembro del Comité Consultivo para Encefalopatías
Espongiforme del Reino Unido, describía así en 1998 el futuro
sobre este tema(26):
"Muchas
cosas han sucedido ya como consecuencia de la aparición de la EEB
en el ganado del Reino Unido. Las medidas adecuadas ya han sido
tomadas para proteger la salud pública y poner fin a la epidemia de
EEB en el ganado vacuno y en otras especies afectadas. Estas medidas
son más rigurosas que nunca. Es difícil adivinar qué podría
haberse hecho para hacer que la epidemia de EEB disminuyera aún más
rápidamente de lo que ha decrecido, aparte de eliminar toda la cabaña
británica, que hubiese sido innecesario y poco práctico. A partir
de ahora la pregunta seguramente será cómo retirar algunas de las
restricciones impuesta a la carne de vaca y a los productos de
vacuno. Las exenciones probablemente se basarán en determinadas
explotaciones (como ha sido el caso de Irlanda del Norte) o
basadas en fechas tras la prohibición total de 1996 sobre el uso de
carne y huesos en la alimentación de cualquier animal de granja en
el Reino Unido. En términos de protección de la salud pública,
todas las medidas necesarias han sido instauradas. Aún persisten
dos motivos de preocupación: si la EEB existe o no en los rebaños
de ovejas y si el número de casos de vECJ en el Reino Unido será
suficientemente alto como para generar preocupación por una segunda
oleada de transmisiones dentro de la población humana como
consecuencia del uso de sangre y derivados sanguíneos. Con respecto
a lo primero, en el Reino Unido se ha intensificado la detección de
ovejas con enfermedades similares al scrapie y la tipificación de
tipos de priones en los animales afectados. Con respecto a la sangre
y derivados sanguíneos, en el Reino Unido ya se han tomado algunas
medidas relativas al uso de productos sanguíneos, y se está
valorando detalladamente el riesgo en relación con la transfusión
sanguínea".
Información
en Internet
En
español:
•
Torres JM, Brun A, Castilla J, Sánchez-Vizcaíno JM. Enfermedades
Producidas Por Priones. Organización Colegial Veterinaria Española.
http://recol.es/comunidades/veterinaria/eeb/priones/priones.htm
• I
Congreso Virtual Iberoamericano de Neurología: http://neurologia.rediris.es/congreso-1/conferencias/priones-10.html
• Consejo
General de Colegios Veterinarios de España. http://www.colvet.es/
• Información
EEB. Administración General del Estado. http://www.eeb.es/
En inglés:
• Report
of the BSE Inquiry ("Informe Phillips") http://www.bseinquiry.gov.uk/report/index.htm
• The
Official Mad Cow Disease Home Page http://www.mad-cow.org/
• The
Virtual Hospital, U. of Iowa. Parenchymal Infections: Prions. http://www.vh.org/Providers/TeachingFiles/CNSInfDisR2/Text/PInf.CDE.html
• Moser
M. Biology and Pathology of Prion Diseases. http://www.prionics.ch/review_e.html
• bmj.com
Collected Resources: New Variant Creutzfeld-Jakob Disease/BSE/Mad
Cow Disease. http://www.bmj.com/cgi/collection/mad_cow
• Prion
Diseases. Dr Shaun Heaphy. Universidad de Leicester. http://www-micro.msb.le.ac.uk/335/Prions.html
• Ministerio
de Agricultura, Pesca y Alimentación del Reino Unido (MAFF) http://www.maff.gov.uk/animalh/bse/index.html
• The
UK Creutzfeldt-Jakob Disease Surveillance Unit http://www.cjd.ed.ac.uk/index.htm
• OMIM,
NCBI. http://www.ncbi.nlm.nih.gov/htbin-post/Omim/dispmim?123400
• Departamento
de Agricultura. Estados Unidos de América. http://www.aphis.usda.gov/oa/bse/
• University
of Illinois at Urbana-Champaign. BSE -- Bovine Spongiform
Encephalopathy ("Mad Cow Disease"). http://w3.aces.uiuc.edu/AnSci/BSE/
• Report
of a WHO Consultation on Clinical and Neuropathological
Characteristics of the New Variant of CJD and Other Human and Animal
Transmissible Spongiform Encephalopathies. Geneva, Switzerland 14 to
16 May 1996 http://www.oms.ch/emc-documents/tse/docs/whoemczoo961.html
• Dealler
S. Information Concerning BSE for the Scientific World. http://sparc.airtime.co.uk/bse/vCJDstats.htm
• Webpath.
CNS Degenerative Diseases http://medstat.med.utah.edu/WebPath/TUTORIAL/CNS/CNSDG.html
•
Nottingham University. The Pathology of Prion Disease
http://www.nottingham.ac.uk/~mbzmail/students/neurodisorders/prionpath.html
Bibliografía
consultada
1
Whitley
RJ, MacDonald N, Asher DM, and the Committee on Infectious Diseases.
Technical Report: Transmissible Spongiform Encephalopathies: A
Review for Pediatricians (T109906). Pediatrics 2000; 106:1160-1165.
Disponible en: http://www.aap.org/policy/t109906.html
2 Detwiler
LA, Rubenstein R. Bovine spongiform encephalopathy: an overview.
ASAIO J 2000; 46: S73-9.
3 Haltia,
M. Human prion diseases. Ann Med 2000; 32: 493-500.
4 Mastrianni
JA, Roos RP. The Prion Diseases. Sem Neurology 2000;
20:337-352. Disponible en: http://www.medscape.com/thieme/SN/2000/v20.n03/sin2003.01.mast/sin2003.01.mast-01.html
5 Polo
JM. Historia y clasificación de enfermedades por priones en
humanos. Rev Neurol 2000; 31: 137-41.
6 Alom
J. Clínica de las enfermedades priónicas humanas. Conferencia en
el Primer Congreso Virtual Iberoamericano de Neurología. http://www.uninet.edu/neurocon/congreso-1/conferencias/priones-5.html
7
Wilesmith JW, Hoinville LJ, Ryan JBM, Sayers AR. Bovine spongiform
encephalopathy: aspects of the clinical picture and analyses of
possible changes. Vet Rec 1992;130:197-201.
8 Bateman
D, Hilton D, Love S, Zeidelr M, Beck J, Collinge J. Sporadic CJD in
a 18-year-old in the UK. Lancet 1995; 346: 1155
9
Britton TC, Sarraj SA, Shaw C, Campbell T, Collinge J. Sporadic CJD
in a 16-year-old in the UK. Lancet 1995; 346: 1155
10
Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K,
Alperovitch A, Poser S, Pocchiari M, Hofman A, Smith PG. A new
variant of CJD in the UK. Lancet 1996; 347: 921-925
11
Whitley RJ, MacDonald N, Asher DM. Technical transmissible
spongiform encephalopathies: A review for pediatricians.
Pediatrics 2000; 106: 1160-5.
12
Will RG. The transmision of prions to humans. Acta Paediatr Suppl
1999; 88: 28-21.
13 Prusiner
SB. Novel proteinacious infectious particles cause scrapie. Science
1982; 216: 136-144.
14
Alper T, Cramp WA, Haig DA, Clarke MC. Does the agent of scrapie
replicate without nucleic acid? Nature 1967;214: 764-766
15
Oesch B, Westaway D, Wälchli M, et al. A cellular gene encodes
scrapie PrP 27-30 protein. Cell 1985;40:735-746
16
Lopez Garcia R, Zahn R, Riek R, Wuthrich K. NMR structure of the
bovine prion protein. Proc Natl Acad Sci USA 2000; 97: 8334-9.
17
Glatzel M, Aguzzi A. Peripheral pathogenesis of prion diseases.
Microbes Infect 2000; 2: 613-9.
18
Büeler H, Aguzzi A, Sailer A, et al. Mice devoid of PrP are
resistant to scrapie. Cell 1993;73:1339- 1347
19
Telling GC, Scott M, Mastrianni J, et al. Prion propagation in mice
expressing human and chimeric PrP transgenes
implicates the interaction of cellular PrP with another protein.
Cell 1995;83:79-90
20
Liao Y-C, Lebo RV, Clawson GA, Smuckler EA. Human prion protein
cDNA: molecular cloning, chromosomal mapping, and biological
implication. Science 1986;233:364-367.
21
Moore RC, Lee IY, Silverman GL, et al. Ataxia in prion protein
(PrP)-deficient mice is associated with upregulation of the
novel PrP-like protein doppel. J Mol Biol 1999;292: 797-817.
22
Collinge J, Sidle KCL, Meads J, Ironside J, Hill AF. Molecular
analysis of prion strain variation and the etiology of new
variant CJD. Nature 1996: 383: 685-690.
23
Hill AF, Zeidler M, Ironside J, Collinge J. Diagnosis of new variant
Creutzfeldt-Jakob diseases by tonsil biopsy. Lancet
1997;349:99-100.
24
Gonzalo I. Cuadrado N. Técnicas de Inmunohistoquímica en las
Enfermedades por Priones. Conferencia en el Primer Congreso
Virtual Iberoamericano de Neurología. http://www.uninet.edu/neurocon/congreso-1/conferencias/priones-10.html
25
Gasset M, Westaway D. Los Priones y su Biología. Conferencia en el
Primer Congreso Virtual Iberoamericano de Neurología.
http://www.uninet.edu/neurocon/congreso-1/conferencias/priones-1.html
26
Pattison J. The Emergence of Bovine Spongiform Encephalopathy and
Related Diseases. Emerging Infectious Diseases 1998;
4:390-394. Disponible en: http://www.medscape.com/govmt/CDC/EID/1998/v04.n03/e0403.06.patt/e0403.06.patt.01.html
27
Wells GAH, Scott AC, Johnson CT, et al. A novel progressive
spongiform encephalopathy in cattle.
Vet Rec 1987;121: 419-420
28
Comisión Europea. Nombre de cas d'Encéphalopathie Spongiforme
Bovine. http://europa.eu.int/comm/food/fs/bse/bse26_fr.pdf
29
The European and Allied Countries Collaborative Study Group of CJD
(EuroCJD) http://www.eurocjd.ed.ac.uk/euroindex.htm
30
The UK CJD Surveillance Unit (1998). http://www.cjd.ed.ac.uk/
31
Will RG, Cousens SN, Farrington CP, Smith PG, Knight RSG, Ironside
JW. Deaths from variant Creutzfeldt-Jakob disease. The Lancet
1999; 353: 979.
32
Heras Pérez, J.A. Nueva variante de la Enfermedad de
Creutzfeldt-Jakob: primer caso diagnosticado en España.
Conferencia en el Primer Congreso Virtual Iberoamericano de
Neurología. http://www.uninet.edu/neurocon/congreso-1/conferencias/priones-6.html
33
Dealler S. Information Concerning BSE for the Scientific World.
34
Haik S, Brandel JP, Sazdovitch V, et al. Dementia with Lewy bodies
in a neuropathologic series of suspected Creutzfeldt-Jakob
disease. Neurology 2000; 55: 1401-4.
35
Lemstra AW, van Meegen MT, Vreyling JP, et al . 14-3-3 Testing in
Diagnosing Creutzfeldt-Jacob Disease: A Prospective Study in
112 Patients. Neurology. 2000;55:514-616
36
Schroter A, Zerr I, Henkel K, Tschampa HJ, Finkenstaedt M, Poser S.
Magnetic resonance imaging in the clinical diagnosis of
Creutzfeldt-Jakob disease. Arch Neurol 2000; 57: 1751-7.
37
Cambier DM, Kantarci K, Worrell GA, et al. Lateralized and focal
clinical, EEG and MRI FLAIR sequence abnormalities in
Creutzfeldt-Jakob disease. Neurology. 2000;54(suppl 3):A100.
Abstract P02.019.
38
World Health Organisation. Report of a WHO consultation on clinical
and neuropathological characteristics of the new variant of
CJD and other human and animal transmissible spongiform
encephalopathies. Geneva: WHO, 1996. (WHO/EMC/ZOO/96:1.)
39
Baumbach G. Infectious Diseases of the Central Nervous System.
Parenchymal Infections: Prions. Department of Pathology,
University of Iowa College of Medicine. Virtual Hospital. http://www.vh.org/Providers/TeachingFiles/CNSInfDisR2/Text/PInf.CDE.html
40
Cuillé J, Chelle PL. Experimental transmission of trembling to the
goat. C R Seances Acad Sci 1939;208:1058-1060
41
Gajdusek DC, Gibbs CJ Jr, Alpers M. Experimental transmission of a
kuru-like syndrome to chimpanzees. Nature 1966;209: 794-796.
42
Gibbs CJ Jr, Gajdusek DC, Asher DM, et al. Creutzfeldt-Jakob disease
(spongiform encephalopathy): transmission to the chimpanzee.
Science 1968;161:388-389.
43
Knox B. Consumer perception and understanding of risk from food. Br
Med Bull 2000; 56: 97-109.
44
Litjen Tan L, Williams MA, Khan MK, Champion HC, Nielsen NH. Risk of
Transmission of Bovine Spongiform Encephalopathy to Humans in
the United States. Report of the Council on Scientific
Affairs. JAMA. 1999;281:2330-2339. Disponible en: http://jama.ama-assn.org/issues/v281n24/abs/jcn90000.html
45
Escudero
Torrella J. Cronología de la Nueva Variante de la Enfermedad de
Creutzfeldt-Jakob. Conferencia en el Primer Congreso Virtual
Iberoamericano de Neurología. http://www.uninet.edu/neurocon/congreso-1/conferencias/priones-4.html
46
Brown P, Cervenakova L, McShane LM, Barber P, Rubenstein R, Drohan
WN. Further studies of blood infectivity in an experimental
model of transmissible spongiform encephalopathy, with an
explanation of why blood components do not transmit
Creutzfeldt-Jakob disease in humans. Transfusion 1999; 39:
1169-78.
47
Rossi F, Legras JF. Viral safety: European and French directives.
Ann Med Interne (Paris) 2000: 151 Suppl 1: 1S55-61.
48 Foster
PR. Prions and blood products. Ann Med 2000; 32: 501-13.
49
Dobson R. Scientists show that vCJD can be transmitted through blood
transfusion. BMJ 2000;321:726. Disponible en: http://www.bmj.com/cgi/content/full/321/7263/726/e
50
Antloga K, Meszaros J, Malchesky PS, McDonnell GE. Prion disease and
medical devices. ASAIO J 2000; 46: S69-72.
51
Reuters Medical News. Fears of 'Mad Cow Disease' Prompt US to
Consider Tighter Blood Donation Limits. Medcape News: http://www.medscape.com/reuters/prof/2001/01/01.18/20010117publ003.html
52
European Commission . Community legislation on BSE. 8 de enero de
2001. http://europa.eu.int/comm/food/fs/bse/bse15_en.pdf
53
de Pedro J. Futuro y problemática de la Vigilancia Epidemiológica
de la Enfermedad de Creutzfeldt-Jakob. Conferencia en el
Primer Congreso Virtual Iberoamericano de Neurología. http://www.uninet.edu/neurocon/congreso-1/conferencias/priones-15.html
54
Timperley WR. Neuropathology J Clin Pathol 2000; 53:255-265.
Disponible en: http://jcp.bmjjournals.com/cgi/content/full/53/4/255
55
Centers for Disease Control. Public Health Service Recommendations
for the Use of Vaccines Manufactured with Bovine-Derived
Materials. Morbidity & Mortality Weekly Report. MMWR
49(50):1137-1138, 2000. Disponible en: http://www.medscape.com/govmt/CDC/MMWR/2000/12.00/mmwr4950.04/mmwr4950.04.html
56
Reuters Medical News. 11 enero 2001. One Quarter of Feed Makers
Noncompliant With 'Mad Cow Disease' Rules. Medcape News:
http://www.medscape.com/reuters/prof/2001/01/01.12/20010111rglt002.html
57
Ortega Albás, JJ. EEG en la enfermedad de Creutzfeldt-Jakob.
Conferencia en el Primer Congreso Virtual Iberoamericano de Neurología.
http://www.uninet.edu/neurocon/congreso-1/conferencias/priones-9.html